L'oxyde de magnésium MgO est un matériau céramique souvent utilisé en électronique, dans les détecteurs, ou encore dans les panneaux photovoltaïques. À l'intérieur de la Terre, lorsqu'il se forme naturellement on le nomme périclase -il contient alors souvent une proportion importante de fer, et on le nomme ferropériclase (Mg,Fe)O. Il s'agit d'un des minéraux les plus abondants du manteau inférieur, entre 700 et 2880 km de profondeur.
Dans ces applications, MgO est souvent présent sous forme polycristalline. Cela signifie que les joints entre les grains jouent un grand rôle dans les propriétés fonctionelles du matériau, en particulier sa déformation plastique. Dans le cadre des projets RheoMan et TimeMan, coordonnés par Patrick Cordier, nous avons employé des simulations par ordinateur pour étudier la structure à l'échelle atomique de joints de grains dans MgO, et leur stabilité lorsqu'ils sont soumis à des perturbations.
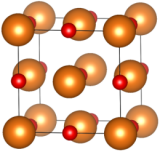
Fig. 1 - Dans la maille élémentaire d'oxyde de magnésium MgO, les ions Mg2+ (représentés ici en orange) forment un réseau cubique à faces centrées (cfc), pénétré par le réseau cfc d'oxygène O2- (en rouge). Si ce cristal se forme naturellement, on le nomme périclase.
Voici 40 ans que les joints de grains dans MgO, et dans d'autres matériaux de même structure, sont étudiés par simulations à l'échelle atomique. En 1974, Kingery propose que les joints de grains dans ces matériaux ioniques doivent avoir une structure similaire à celle des joints dans les métaux, c'est-à-dire une structure compacte comme sur la Fig. 2 à gauche [1].
Dès les années 1980, les simulations atomistiques deviennent possibles, et des gens les utilisent pour simuler des joints de grains dans des matériaux ioniques. Duffy et Tasker sont parmi les premiers, et ils obtiennent des structures ouvertes, dans lesquelles les deux grains sont séparés par du vide, comme au milieu de la Fig. 2 [2]. Selon eux, dans la structure compacte de Kingery, des ions de même nature se trouvent en vis-à-vis à l'interface (voir les cercles blancs de part et d'autre des unités structurelles en vert), leur répulsion électrostatique est donc trop importante. Au contraire, dans la structure ouverte, ce sont des ions de nature différente qui se retrouvent en vis-à-vis, ce qui stabilise la structure (voir unités structurelles rouges). Suite à ces travaux, tous les groupes ayant mené des simulations à l'aide de potentiels inter-atomiques, ou de méthodes ab initio, ont essentiellement confirmé ce résultat : dans les matériaux ioniques à structure NaCl, les joints de flexion sont ouverts dans leur état fondamental.
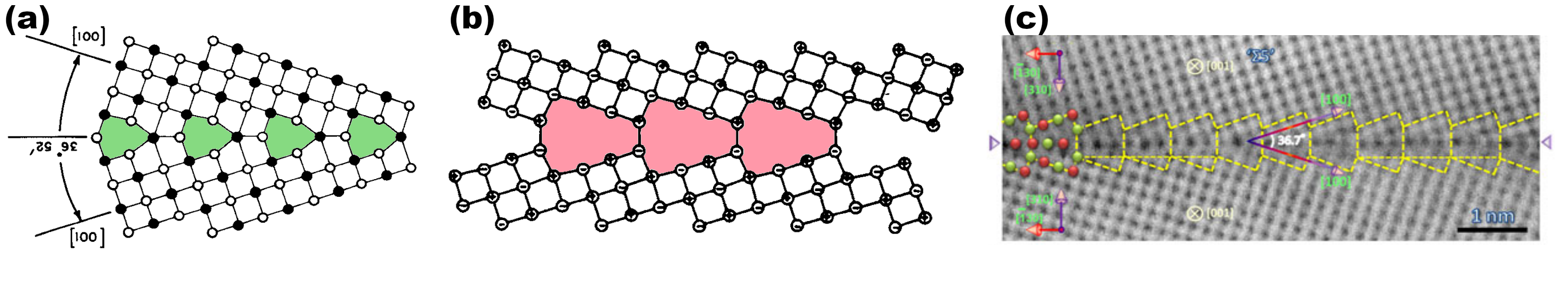
Fig. 2 - Différentes structures, prédites ou observées, d'un joint de grains de flexion Σ5 dans des matériaux ioniques à structure NaCl. (a) En 1974, Kingery propose que les joints de grains dans NaCl sont compacts. Les unités structurelles sont colorées en vert. (b) En 1982, Duffy et Tasker réalisent les premières simulations de joints de grains dans NiO, et prédisent que les joints de grains doivent avoir des structures ouvertes, sans colonne atomique dans le plan du joint (en rouge). Depuis, toutes les simulations (avec potentiels ou ab initio) confirment ce résultat. (c) Le joint Σ5 observé par Saito et al. en 2013, montrant des colonnes atomiques à l'intérieur des unités structurelles : ce joint est compact.
Mais alors, que disent les observations ? Il a longtemps été difficile d'observer des joints de grains avec une résolution atomique. Mais depuis les années 2010, les observations de joints de grains en microscopie à haute résolution se multiplient, comme sur la Fig. 2 à droite. Ces observations montrent toutes des joints de grains fermés, c'est-à-dire des unités structurelles non pas vides, mais contenant des colonnes atomiques dans le plan du joint. Elles semblent donc remettre en cause les structures ouvertes prédites par les simulations. Comment expliquer ces différences ?
Pour débuter, nous avons focalisé notre étude sur l'état fondamental des joints de grains de flexion symétrique. Deux cristaux de MgO, dont les axes [001] sont alignés, sont pivotés d'angles opposés ±α/2 autour de cet axe, puis superposés. Ils se rencontrent ainsi avec le même plan cristallographique {hk0}, où les indices de Miller h et k dépendent de l'angle de rotation initial.
Aux faibles angles (0°<α<20°), les cristaux accomodent la désorientation grâce à des dislocations [010] espacées périodiquement. Cette situation est bien décrite par le modèle de Read et Shockley, et les énergies obtenues par les simulations correspondent bien à celles prédites par ce modèle (Fig. 3). Aux angles plus grands, cette description n'est plus adaptée : la structure atomique ne peut plus être décrite uniquement avec des dislocations, et le modèle de Read et Shockley échoue à prédire les énergies. Les simulations atomistiques indiquent que les joints de grains de fortes désorientations (20°<α<67.4°) ont des énergies assez similaires, formant un plateau à environ 1,7 J/m2.
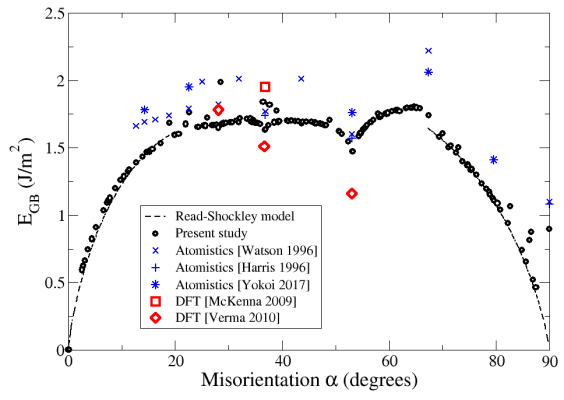
Fig. 3 - Énergie minimale de joints de grains de flexion symétriques dans MgO. Les résultats de notre étude (cercles noirs) sont comparés avec des calculs ab initio (carrés et losanges rouges), des simulations atomistiques (croix et étoiles bleues), et le modèle de Read et Shockley (lignes pointillées).
La désorientation particulière α=67.4° marque une discontinuité dans les énergies des joints de grains, et aussi dans leur structure atomique. Au-delà de cet angle, il devient plus favorable de déplacer les cristaux l'un par rapport à l'autre le long du joint de grain. La raison est la suivante : pour la désorientation α=90°, les deux cristaux ont chacun été tournés de 45° et forment un simple défaut d'empilement ; translater un cristal par rapport à l'autre permet de retrouver le cristal parfait. Pour les angles proches de l'angle droit (67.4°<α<90°), il est donc plus favorable de translater un cristal afin de maximiser le volume de cristal parfait, la désorientation restante étant accomodée par des dislocations de type ½[110]. Ces joints de grains répondent alors à nouveau au modèle de Read et Shockley, qui prédit très bien leurs énergies (Fig. 3).
Comme toutes les autres auparavant, notre étude confirme que dans l'état fondamental, tous les joints de flexion ont une structure ouverte, les deux cristaux de MgO semblant séparés par du vide [3]. Puisque ce résultat surgit de toutes les méthodes de simulations, que ce soit avec des potentiels inter-atomiques ou des méthodes ab initio, l'observation de joints compacts doit trouver une explication ailleurs.
Les joints de grains précédents ont été construits avec deux cristaux de MgO parfaitement alignés, parfaitement stœchiométriques, et sans le moindre défaut sur leurs surfaces initiales. Cela peut être considéré comme un peu trop idéalisé. Dans la suite, nous avons alors considéré l'importance du désordre et de la fraction atomique sur l'état d'un joint de grains.
La fraction atomique peut être définie comme le nombre de « blocs de construction » disponibles à l'interface. Dans notre cas, il s'agit du nombre de paires d'ions Mg-O présentes à l'interface. Afin de le faire varier, nous avons construit des systèmes atomiques contenant un joint de grain Σ5 dans sa complexion ouverte. Le système est dupliqué dans les deux directions du plan du joint, afin de varier la fraction atomique par petits paliers. Après chaque paire Mg-O retirée, une simulation en dynamique moléculaire permet au joint de se réarranger, notamment par migration d'ions dans le plan du joint.
Les résultats sont présentés sur la Fig. 4. Démarrant du joint ouvert, retirer des paires Mg-O fait rapidement augmenter son énergie propre. Cela traduit le fait que cette complexion ouverte n'est pas stable par rapport à une variation de la fraction atomique. Dans le même temps la structure devient rapidement compacte, des atomes allant remplir les unités structurelles à l'intérieur du joint. Une fois compact, le joint est alors relativement insensible à la fraction atomique : retirer davantage de paires Mg-O change bien sûr sa structure, mais n'a qu'un effet modéré sur son énergie et sa compaction. Plus important, le joint reste compact, et ne retourne alors jamais vers une configuration ouverte. Le changement de structure est irréversible.
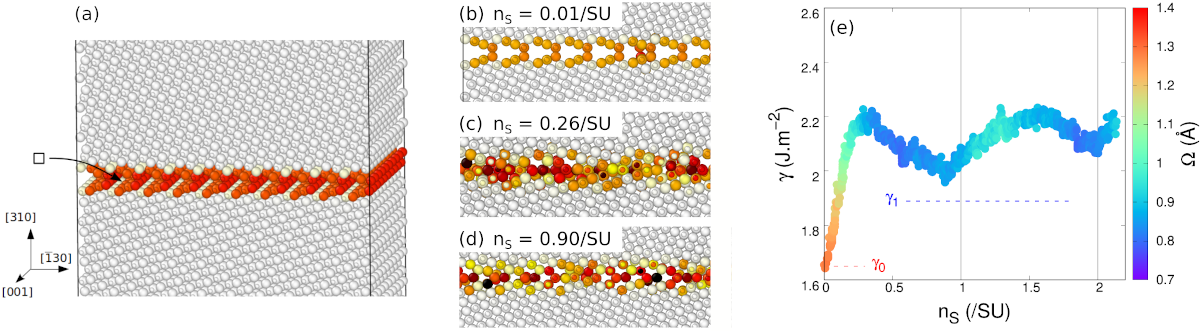
Fig. 4 - Évolution du joint de grain {310}[001] avec sa fraction atomique. (a) Le système est construit avec un joint de grains ouvert et de large surface. Après chaque paire Mg-O retirée, une simulation en dynamique moléculaire permet au joint de grain de se réarranger. (b-d) À mesure que des paires Mg-O sont retirées, le joint devient rapidement compact. (e) L'énergie propre du joint de grain, notée γ, augmente rapidement lorsque des paires Mg-O sont retirées du joint ouvert. Dans le même temps son volume d'excès Ω (code couleur) diminue aussi rapidement, traduisant sa compaction. Par la suite, alors que davantage de paires Mg-O sont retirées, le joint reste compact, son énergie varie relativement peu.
Nous avons réalisé ces simulations sur des joints {410}, {310} et {210} dans MgO, et obtenu dans chaque cas des résultats similaires. Ces résultats ont été publiés dans Acta Materialia [4]
[1] W.D. Kingery, J. Amer. Ceram. Soc. 57 (1974) 1-8. doi: 10.1111/j.1151-2916.1974.tb11350.x
[2] D.M. Duffy, P.W. Tasker, Philos. Mag. A 47 (1983) 817-825. doi: 10.1080/01418618308243121
[3] P. Hirel et al., Phys. Chem. Miner. 46 (2019) 37-49. doi: 10.1007/s00269-018-0985-7
[4] P. Hirel et al., Acta Mater. 240 (2022) 118297. doi: 10.1016/j.actamat.2022.118297
Copyright © Pierre Hirel 2009-2024
