Magnesium oxide MgO is a ceramic material often used in electronics, sensors, or photovoltaic applications. Deep inside the Earth, where it forms naturally, it is called periclase -it then often contains an important proportion of iron, and it is named ferropericlase (Mg,Fe)O. It is one of the most abundant minerals of Earth's lower mantle, between 700 and 2880 km depth.
In all these situations, MgO is often polycristalline. It means that the boundaries between grains play a major role in the functional properties of the material, in particular its plastic deformation. In the framework of the RheoMan and TimeMan projects, coordinated by Patrick Cordier, we employed numerical simulations to study the atomic-scale structure of grain boundaries in MgO, and their stability when they are submitted to perturbations.
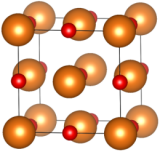
Fig. 1 - In the unit cell of MgO, Mg2+ ions (represented as orange spheres) form a face-centered cubic (fcc) lattice, penetrated by another fcc lattice of oxygen ions O2- (in red). When formed naturally, this crystal is called periclase.
It has now been 40 years since grain boundaries in MgO, and in other materials with the same structure, are studied with atomic-scale simulations. In 1974, Kingery proposes that grain boundaries in such ionic materials probably have an atomic structure similar to those in metals, i.e. a compact structure like the one on the left of Fig. 2 [1].
As soon as the 1980s, atomistic simulations become possible, and people bein using them to simulate grain boundaries in ionic materials. Duffy and Tasker are among the first, and they obtain open structures, in which the two grains are separated by void, like in the middle of Fig. 2 [2]. According to them, in the compact structure of Kingery ions of same nature face each other at the interface (see white circles facing each other in green structural units), therefore their electrostatic repulsion is too great. On the contrary, in the open structure ions of different nature face each other, which stabilizes the structure (see red structural units). Following these publications, each and every group who performed simulations with inter-atomic potentials, or with ab initio method, only confirmed this result: in ionic materials with the rock-salt lattice, tilt grain boundaries have an open structure in their ground state.
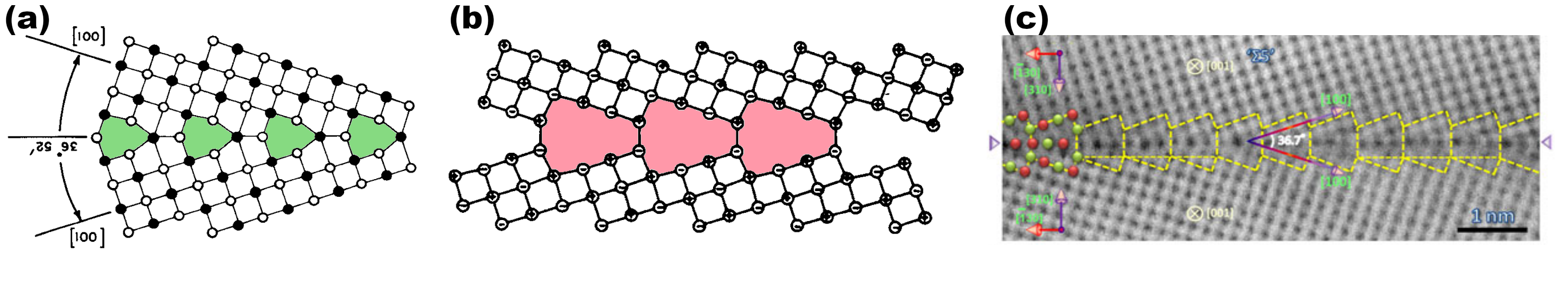
Fig. 2 - Different structures, predicted or observed, of a Σ5 symmetric tilt grain boundary in rock-salt materials. (a) In 1974, Kingery proposes that grain boundaries in NaCl are compact. Structural units are coloured in green. (b) In 1982, Duffy and Tasker perform the first atomistic simulations of grain boundaries in NiO, and predict that they must have open structures, without any atomic column in their vicinity (in red). Since then, all simulations (with inter-atomic potentials or ab initio) confirm this result. (c) The Σ5 grain boundary observed by Saito et al. in 2013, showing atomic columns inside structural units: this grain boundary is compact.
But then, what do observations say? For a long time it has been difficult to observe grain boundaries with an atomic resolution. But since the 2010s, observations of grain boundaries with high-resolution electron microscopy multiply, like the one on the right of Fig. 2. These observations all show compact grain boundaries, i.e. structural units which are not void, but filled with atomic columns. They seem to contradict the open structures predicted by simulations. How can we explain such discrepancies?
To begin, we focused on the ground state of symmetric tilt grain boundaries (STGB). Two MgO single crystals which [001] axes are aligned, are rotated by opposite angles ±α/2 around that axis, cut, and then stacked. They meet with equivalent crystallographic planes {hk0}, where Miller indices h and k depend on the chosen misorientation angle α.
At low angles (0°<α<20°), the crystals accomodate the misorientation by forming [010] dislocations, which are regularly spaced. This situation is well described by the elastic model by Read and Shockley, and the energies obtained in atomistic simulations fit quite well with those predicted by this model (Fig. 2). At higher angles, this description is not suited any longer: the atomic structure cannot be described solely in terms of dislocations anymore, and the Read-Shockley model fails to predict the energies. Atomistic simulations indicate that for such high misorientations (20°<α<67.4°), grain boundaries have similar energies about 1,7 J/m2.
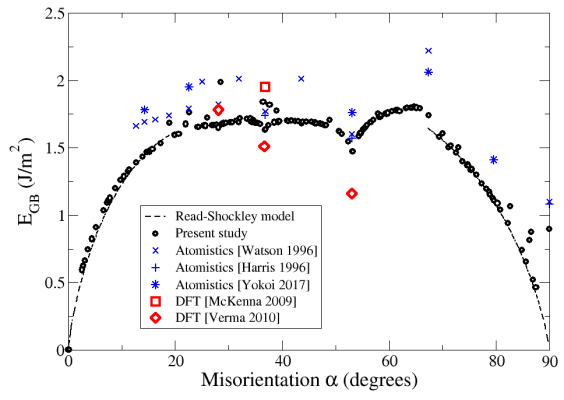
Fig. 2 - Minimum energies of symmetric tilt grain boundaries in MgO. Our results (black circles) are compared with ab initio calculations (red squares and diamonds), other atomistic simulations (blue crosses and stars), and the Read-Shockley model (dashed lines).
The specific misorientation α=67.4° marks a discontinuity in the GB energies, and also in their atomic structure. Beyond this misorientation angle, it becomes more favorable to shift one crystal with respect to the other along the grain boundary. The reason for this is the following: for the misorientation α=90°, the two crystals are both rotated by ±45° and form a simple stacking fault; shifting one crystal with respect to the other allows to restore the perfect crystal. For angles close to the right angle (67.4°<α<90°), it is therefore more favorable to shift one crystal in order to maximize the volume of perfect crystal, the small remaining misorientation being accomodated by dislocations with a ½[110] Burgers vector. Those grain boundaries are then well described by the Read-Shockley model, which predicts their energies with a good accuracy (Fig. 2).
Like all others before, our study shows that in their ground state, all symmetric tilt grain boundaries have an open structure, the two MgO crystals being separated by void [3]. Since this result arises commonly from all simulations, either using empirical potentials or ab initio methods, it means that the explanation for the observation of compact grain boundaries must be found elsewhere.
The previous grain boundaries were constructed from two MgO crystals that were perfectly aligned, perfectly stoichiometric, and without any defect on their initial surfaces. This can be considered as too idealized. In the following, we investigated the effect of disorder and atomic fraction on the state of grain boundaries.
Atomic fraction can be defined as the number of "building blocks" available at the interface. In our case, it is the number of pairs of Mg-O ions present at the interface. To vary it, we constructed atomistic systems containing a Σ5 grain boundary in its open complexion. The system is duplicated in both directions in the plane of the boundary, so as to vary the atomic fractions by small increments. After each Mg-O pair removed, a molecular dynamics simulation allows the atoms to rearrange at the boundary, in particular by migrating in the plane of the grain boundary.
Results are presented in Fig. 4. Starting from the open grain boundary, removing Mg-O pairs rapidly increases its intrinsic energy. This means that this open complexion is unstable with respect to variations of the atomic fraction. At the same time, the structure becomes more compact, and atoms fill in the voids inside the structural units. Once compact, the grain boundary is relatively insensitive to further variation of the atomic fraction: removing more Mg-O pairs changes its structure, but has only a mild effect on its energy ad compaction. More importantly, the grain boundary remains compact, and never goes back to an open configuration. The change of structure is irreversible.
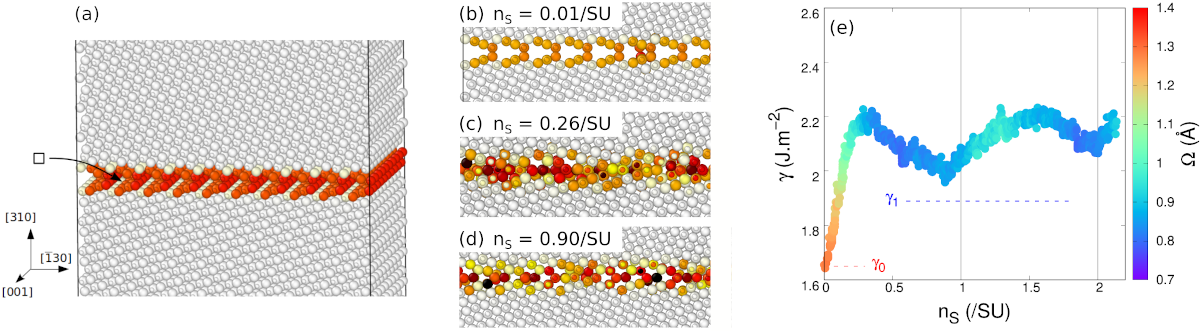
Fig. 4 - Evolution of the {310}[001] grain boundary with atomic fraction. (a) The system is constructed with an open grain boundary with a large surface. After each Mg-O pair removed, a molecular dynamics simulation allows for atomic rearrangements. (b-d) As Mg-O pairs are removed, the grain boundary rapidly becomes compact. (e) The grain boundary intrinsic energy, noted γ, increases rapidly as Mg-O pairs are removed. Meanwhile its excess volume Ω (colour code) decreases also rapidly, indicating its compaction. After that, as more Mg-O pairs are removed, the grain boundary remains compact, its energy varying more slowly.
We performed such simulations on {410}, {310} and {210} symmetric tilt grain boundaries in MgO, and obtained in each case similar results. This study was published in Acta Materialia [4]
[1] W.D. Kingery, J. Amer. Ceram. Soc. 57 (1974) 1-8. doi: 10.1111/j.1151-2916.1974.tb11350.x
[2] D.M. Duffy, P.W. Tasker, Philos. Mag. A 47 (1983) 817-825. doi: 10.1080/01418618308243121
[3] P. Hirel et al., Phys. Chem. Miner. 46 (2019) 37-49. doi: 10.1007/s00269-018-0985-7
[4] P. Hirel et al., Acta Mater. 240 (2022) 118297. doi: 10.1016/j.actamat.2022.118297
Copyright © Pierre Hirel 2009-2026
